Wilson disease is a rare, hereditary genetic ailment that impairs the body’s capacity to metabolize copper, resulting in its toxic buildup in essential organs such as the liver, brain, and eyes. Wilson disease results from mutations in the ATP7B gene, which facilitates the transport of excess copper from the liver to the bile for excretion from the body. When this process malfunctions, copper accumulates in organs, resulting in extensive harm. Although Wilson illness may be asymptomatic for years, its manifestations might vary from hepatic issues in childhood to neurological and mental symptoms in adolescence or adulthood. In the absence of treatment, the disease deteriorates steadily, resulting in hepatic failure, neurological impairment, and further health issues.
Clinical Description, classification and symptoms
| Classification Level | Disorder |
| Prevalence | 1-9 / 100,000 |
| Age of Onset | Adolescent, Adult, Childhood, Elderly |
| Inheritance | Autosomal Recessive |
The clinical presentation of Wilson disease is highly variable, even among affected families. The age of onset can differ significantly, with some individuals remaining asymptomatic for decades, while others show symptoms as early as 3 to 5 years old. Typically, symptoms begin to appear by age 40, although late-onset cases in individuals over 50 have also been documented. The clinical manifestations depend on factors such as gender and age. In children, liver-related symptoms are usually the first to develop, often by the age of 10, and these are commonly accompanied by liver damage such as hepatomegaly, hepatitis, liver failure, or cirrhosis. Neurological symptoms like dystonia, tremors, difficulty speaking, coordination problems, and gait issues may occur alongside liver symptoms or may be the initial signs of the disease. Isolated psychiatric disorders, including depression, phobias, personality changes, or emotional instability, are rare but may be seen alongside liver or neurological issues. Additional symptoms can include acute hemolytic episodes, delayed puberty, amenorrhea, recurrent miscarriages, copper deposits in the eyes (Kayser-Fleischer rings), bone pain, joint pain, osteoporosis, heart arrhythmias, cardiomyopathy, hematuria, nephrotic syndrome, kidney stones, and, in rare cases, hepatocellular carcinoma.
Diagnosis and Treatment
The diagnosis relies on clinical features and abnormal lab results, such as elevated liver enzymes, thrombocytopenia, low serum ceruloplasmin, and elevated urinary copper excretion. A liver biopsy may be used to detect high copper levels, and genetic testing is used to confirm the diagnosis in approximately 98% of cases. Family screening can identify 20% of additional cases.
For liver symptoms, differential diagnoses include acute or chronic hepatopathy, such as viral or autoimmune hepatitis, non-alcoholic steatohepatitis, primary sclerosing cholangitis, primary biliary cirrhosis, and alpha-1-antitrypsin deficiency. For neurological symptoms, conditions like essential tremor, early-onset Parkinson’s disease, and dystonia must be considered. Wilson disease is inherited in an autosomal recessive pattern, and genetic counseling is recommended for at-risk couples, as there is a 25% chance of having an affected child if both parents are carriers.
The prognosis of Wilson Disease depends on early diagnosis, timely treatment, and adherence to the treatment plan. Since treatment for Wilson disease is ongoing for life, prompt intervention is crucial, as delays in treatment may lead to irreversible damage. Treatment focuses on establishing a negative copper balance through lifelong use of chelating agents (D-penicillamine, trientine) or zinc salts. Treatment improves symptoms over time, but its effectiveness depends on strict lifelong adherence. Restricting copper-rich foods may also help. In cases of acute liver failure or decompensated cirrhosis, liver transplantation is recommended. Chelation therapy medications approved for treating Wilson disease include penicillamine (Cuprimine® and Depen®), trientine dihydrochloride (Syprine®), and trientine tetrahydrochloride (Cuvrior™). These drugs work by binding to copper, which increases its excretion through urine. Zinc acetate, sold as Galzin™ in the U.S. and Wilzin® in Europe, is another treatment option. Zinc helps by stimulating metallothionein production and blocking the absorption of copper in the gut. This both depletes accumulated copper and prevents its buildup. Zinc is primarily used for long-term maintenance, treating asymptomatic patients, and can be used alongside chelation therapy. It has been proven effective through over 40 years of use in the U.S. and Europe, and one of its key benefits is its minimal side effects.
Also, UX701 is an investigational gene therapy using AAV9 technology, aimed at delivering stable expression of the ATP7B copper transporter with a single intravenous infusion. Preclinical studies have shown that UX701 can normalize copper trafficking and excretion in the body. It has received Orphan Drug Designation in both the United States and European Union and fast track designation in the U.S. for its potential in treating Wilson disease.
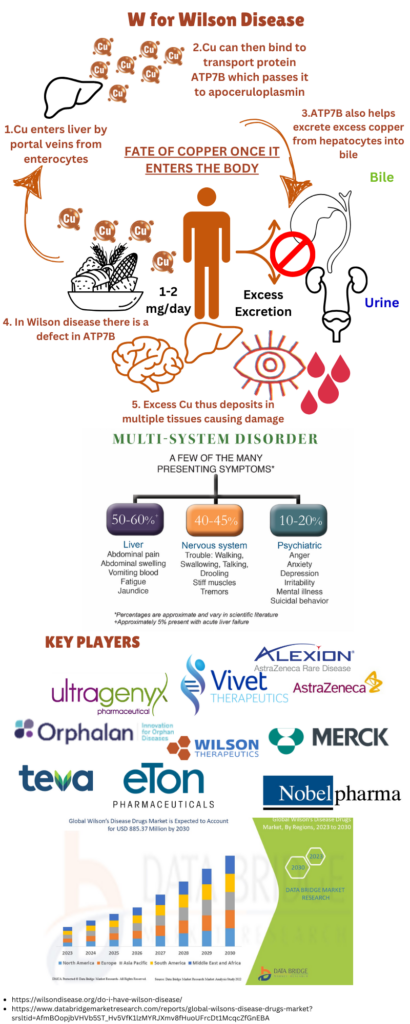
Image: An Overview of Wilson Disease; Key Players and Global Market involved in Treatment of Wilson Disease
If untreated, Wilson disease is progressive and can be life threatening, but with appropriate treatment, long-term outcomes are very favorable. The main goal of treatment is to remove excess copper buildup and prevent its reaccumulation. The Wilson’s Disease diagnostic market, The market was valued at USD 527 million in 2022 and is expected to reach USD 885.37 million by 2030.
For more on rare diseases, stay tuned for next installment of Care for Rare. Meanwhile, click here for more available resources and support groups for Wilson Disease.
Dr. Malini Gupta, Ph.D,
Sources
- https://www.marketreportanalytics.com/reports/wilsons-disease-diagnostic-25573#summary
- https://rarediseases.org/rare-diseases/wilson-disease/
- https://www.orpha.net/en/disease/detail/905
- Figure idea from – https://www.osmosis.org/video/Wilson_disease
Disclaimer
The editors take care to share authentic information. In case of any discrepancies please write to newsletter@medness.org
The sponsors do not have any influence on the nature or kind of the news/analysis reported in MedNess. The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of MedNess. Examples of analysis performed within this article are only examples. They should not be utilized in real-world analytic products as they are based only on very limited and dated open-source information. Assumptions made within the analysis are not reflective of the position of anyone volunteering or working for MedNess. This blog is strictly for news and information. It does not provide medical advice, diagnosis or treatment nor investment suggestions. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
MedNess is a part of STEMPeers® which is a 501(c)(3) organization registered in PA as PhD Career Support Group. The organization helps create a growing network of STEM scientists that is involved in peer-to-peer mentoring and support.

