I-cell disease, also known as Mucolipidosis type II, is a severe rare lysosomal storage disorder caused by mutations in the GNPTAB gene located on chromosome 12q23.2. This gene encodes the alpha and beta subunits of N-acetylglucosamine-1-phosphotransferase, which plays a critical role in processing and packaging lysosomal enzymes by tagging them with mannose-6-phosphate in the Golgi apparatus. Mutations in this gene lead to a failure of mannose-6-phosphate (MP6) synthesis, the marker on the oligomannosyl type glycan side chains of lysosomal enzymes, which targets the enzymes to the lysosomes. In the absence of the MP6 signal, the enzymes are sorted erroneously into the extracellular space, resulting in the accumulation of the enzymes in the tissues. Consequently, cells accumulate mucolipids and mucopolysaccharides, resulting in dysfunction across various tissues. MLII is the most severe form among the mucolipidoses, characterized by a complete deficiency of GlcNAc phosphotransferase activity. Mucolipidosis II (ML II) and Mucolipidosis III (ML III) are variations of the same disease caused by deficiencies in a targeting enzyme. ML II is characterized by more severe clinical features, while ML III presents with attenuated or less severe symptoms. Both conditions stem from mutations affecting the same metabolic pathway, leading to impairments in lysosomal enzyme processing and function. May 15th is observed as the Mucopolysachharidosis and Mucolipidosis awareness day.
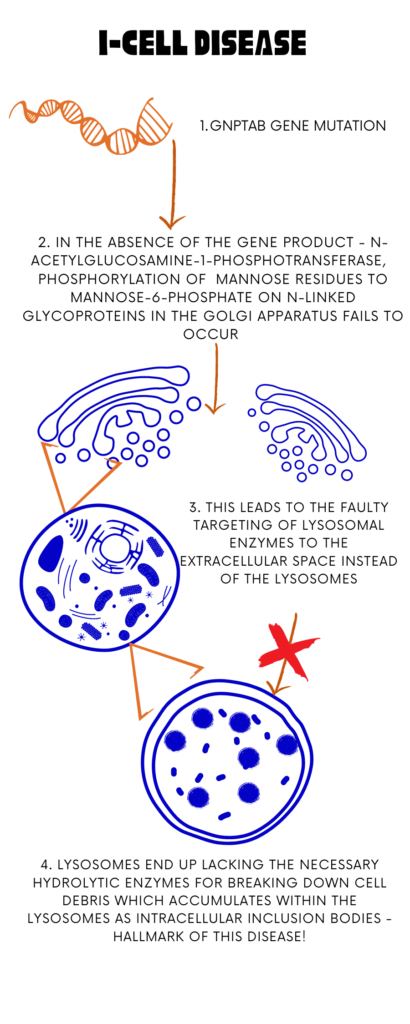
Image I : I- Cell Disease- An Overview; Image Source: Malini Gupta
Clinical Description
| Classification Level | Disorder |
| Prevalence | <1/1000000 |
| Age of Onset | Prenatal, Antenatal |
| Inheritance | Autosomal recessive |
- Patients with MLII typically exhibit profound clinical manifestations that begin either before birth or shortly after birth.
- These include distinct facial dysmorphism, cardiac complications, respiratory problems, dysostosis multiplex (abnormal skeletal development), severe growth retardation, and significant delays in both cognitive and motor development.
- The median age of diagnosis is approximately 0.7 years, with a median survival of around 5.0 years, and death often occurs before the age of 10 due to complications related to heart and lung function.
- Other notable features may include clubfeet, hypotonia (low muscle tone), and significant delays in global development, all of which progressively worsen over time.
- Gradual development of spinal cord compression is also observed. Growth typically ceases within the first two years of life, resulting in joint contractures that severely limit mobility and prevent many patients from walking.
- Cardiac involvement is frequent and often includes thickening and functional insufficiency of the mitral or aortic valves. Breathing difficulties arise due to progressive narrowing of the airways, thickening of mucosal tissues, and stiffening of connective tissues throughout the body, contributing to cardiorespiratory insufficiency.
Diagnosis and Current Treatment
Currently, there is no cure for mucolipidosis II or I-cell disease, and treatment focuses on supportive care. Strategies include interactive programs to stimulate cognitive development, low-impact therapies such as aqua therapy, and occupational or speech therapy Experimental approaches such as hematopoietic stem cell transplantation (HSCT), which has shown some promise in other lysosomal storage disorders, have been attempted in a limited number of MLII cases. However, outcomes vary widely, with only a few reported instances of potential benefit from HSCT. Diagnosis of MLII is confirmed through biochemical analysis, particularly by observing a significant increase (typically at least tenfold) in plasma levels of lysosomal enzymes such as arylsulfatase A and β-glucuronidase. Genetic testing targeting the GNPTAB gene is essential to definitively establish the diagnosis. Inheritance is autosomal recessive, prompting genetic counseling for at-risk couples who both carry disease-causing mutations. Each pregnancy carries a 25% risk of having an affected child.
For more on the latest curatives for this disease, stay tuned for I for I-Cell Disease part II. Meanwhile, click here for more available resources and support groups for I-Cell Disease.
Dr. Malini Gupta, Ph.D.
Sources
- https://www.orpha.net/en/disease/detail/576
- https://rarediseases.org/rare-diseases/i-cell-disease/
- https://www.frontiersin.org/journals/pediatrics/articles/10.3389/fped.2023.1199489/full
- https://mpssociety.org/learn-about-mps/diseases/ml-ii-ml-iii/
Disclaimer
The editors take care to share authentic information. In case of any discrepancies please write to newsletter@medness.org
The sponsors do not have any influence on the nature or kind of the news/analysis reported in MedNess. The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of MedNess. Examples of analysis performed within this article are only examples. They should not be utilized in real-world analytic products as they are based only on very limited and dated open-source information. Assumptions made within the analysis are not reflective of the position of anyone volunteering or working for MedNess. This blog is strictly for news and information. It does not provide medical advice, diagnosis or treatment nor investment suggestions. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
MedNess is a part of STEMPeers® which is a 501(c)(3) organization registered in PA as PhD Career Support Group. The organization helps create a growing network of STEM scientists that is involved in peer-to-peer mentoring and support.

