Gaucher disease is a rare, genetic metabolic disorder where glucocerebrosidase deficiency lead to accumulation of fat-laden “Gaucher cells” in areas like the spleen, liver and bone marrow. Gaucher cells are normal macrophages which become full of unprocessed glucocerebroside and accumulate in the organs causing dysfunction and inflammation. This is the second most severe type of lysosomal storage disorder (LSD) after Fabry disease. Lysosomes serve as the primary digestive compartments within cells, breaking down complex carbohydrates and fats. Characteristic of Gaucher disease, is the abnormal accumulation of glucose-containing fats called glycolipids in the body. This lipid buildup leads to the diverse symptoms characteristic of LSDs. There is a lot of variability of symptoms and physical findings between patients with Gaucher Disease; where some are asymptomatic while others have severe. Three distinct types of Gaucher disease have been identified and are distinguished by the absence, presence or the extent of, neurological complications. Gaucher disease can occur in up to 1 in 40,000 live births in the general population. Individuals will get Gaucher Disease if both parents are carriers. October is celebrated as the Gaucher disease awareness month.
| Classification Level | Disorder |
| Prevalence | 1-9/100,000 |
| Age of Onset | All ages |
| Inheritance | Autosomal Recessive |
Clinical Description
- Gaucher disease commonly presents with an enlarged liver and/or spleen (hepatosplenomegaly),
- reduced levels of red blood cells (anemia)
- decreased platelet counts (thrombocytopenia), and skeletal irregularities. Thrombocytopenia can lead to bleeding issues due to decreased platelets, crucial for clotting.
- There are three recognized types of Gaucher disease, categorized by the presence or absence and severity of neurological complications. Inheritance of all three forms follows an autosomal recessive pattern.
Diagnosis and Current Treatment
Gaucher disease is categorized into three types based on the presence or absence of early-onset brain involvement:
Gaucher disease type 1: This form is predominant in western nations, constituting about 95 percent of cases there. Symptoms include enlarged spleen and liver, bone issues, and fatigue. Brain development remains unaffected. Gaucher disease type 1 is treatable.
Gaucher disease type 2: This type is exceedingly rare and characterized by severe neurological abnormalities, primarily affecting the brain stem. Unfortunately, it typically results in fatality within the first two years of life due to irreversible brain damage, and currently lacks effective treatment.
Gaucher disease type 3: While uncommon in the United States and Europe, it is the most prevalent form of Gaucher disease globally. Gaucher disease type 3 presents symptoms like type 1 along with some neurological involvement. While patients often have a shortened lifespan, some can live into their 50s with proper treatment.
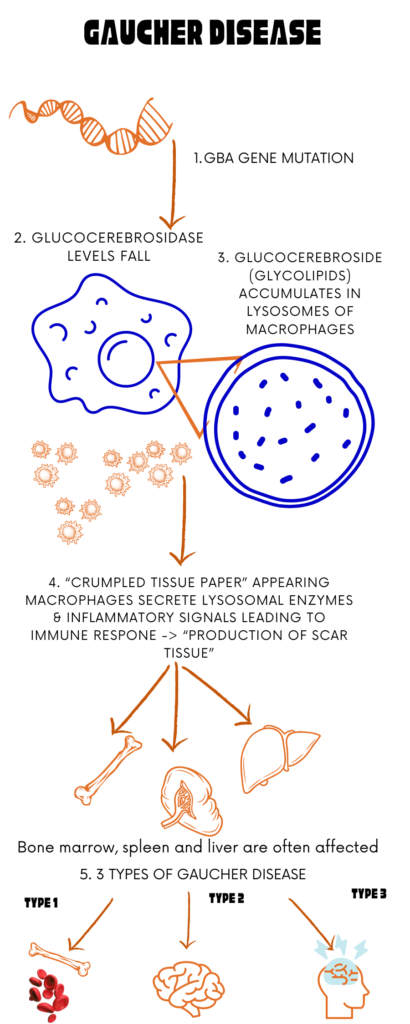
Image: Types of Gaucher Disease; Image Source: Malini Gupta
Gaucher disease type 1, prevalent in western nations, is manageable. Even non-neurological symptoms in type 3, the most widespread globally, are treatable. For type 3 patients without severe brain complications, treatment can alleviate symptoms and enhance well-being. Therapeutic options comprise enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). Collaborating with a specialist in Gaucher disease is crucial for ongoing health monitoring and medication adjustments when needed. Apart from ERT and SRT, which tackle enzyme deficiency and glucocerebroside buildup, additional treatments may be necessary to manage symptoms and complications of Gaucher disease. These treatments encompass: Blood transfusions to address severe anemia and bleeding, medications prescribed for bone pain and osteoporosis, orthopedic surgical interventions like joint replacement to alleviate pain and repair damaged joints.
For more on the latest curatives for this disease, stay tuned for G for Gaucher Disease part II. Meanwhile, click here for more available resources and support groups for Gaucher Disease.
Dr. Malini Gupta, Ph.D.
Sources

