Fanconi Anemia (FA) is a rare autosomal recessive disorder caused due to mutations in any of the known 23 genes that play a role in the DNA repair pathway. Defect in the DNA repair in all cells of the body results in accumulation of errors in these cells over time, thus making people living with FA vulnerable to developing bone marrow failure and cancer in addition to many other systemic issues. More than 90% of patients with FA have mutations in the FANCA, FANCC, FANCG, or FANCD2 genes. Mutations in genes that play an upstream role in the FA-core complex are linked to classic FA. Conversely, downstream variants, such as those in BRCA2, are associated with a significantly increased risk of developing solid tumors during infancy and early childhood. FA affects both genders equally. Birth abnormalities, poor hematopoiesis, a higher risk of acute myeloid leukemia, myelodysplastic syndrome, and malignancies are the major characteristics of FA. May 1st is celebrated as Fanconi Anemia Awareness Day.
| Classification Level | Disorder |
| Prevalence | 1-9/1000,000 |
| Age of Onset | Childhood |
| Inheritance | Autosomal recessive, X-linked recessive |
Orphanet Description
Individuals with FA are usually smaller than average and may experience extreme fatigue and frequent infections. Early signs of the disease can include nosebleeds or easy bruising. low white or red cell, or platelet count, occasionally, myelodysplasia, acute myeloid leukemia (AML), or squamous cell carcinoma in a young adult is the first indication of FA.
- The first signs of Fanconi anemia (FA) are often non-hematological. Limb anomalies typically affect the extremities, either unilaterally or (usually asymmetrically) bilaterally.
- Minor anomalies can include low birth weight, microcephaly, microphthalmia, common skin pigmentation abnormalities – café-au-lait: frequent hypoplastic thenar eminence.
- Short stature is syndromic and/or associated with endocrinopathies, fertility issues occur frequently in males, highly disturbed in about half of females.
- Diagnosis may be delayed until hematological anomalies appear if congenital malformations are not prominent.
- Bone marrow failure (BMF): Occurs at a median age of 7 years and develops in 90% of patients by 40 years of age with early manifestations including macrocytosis and thrombocytopenia.
- In patients with somatic mosaicism, blood counts may remain normal until the onset of hematological malignancy and high pre-disposition to solid tumors, especially in the head and neck or anogenital regions.
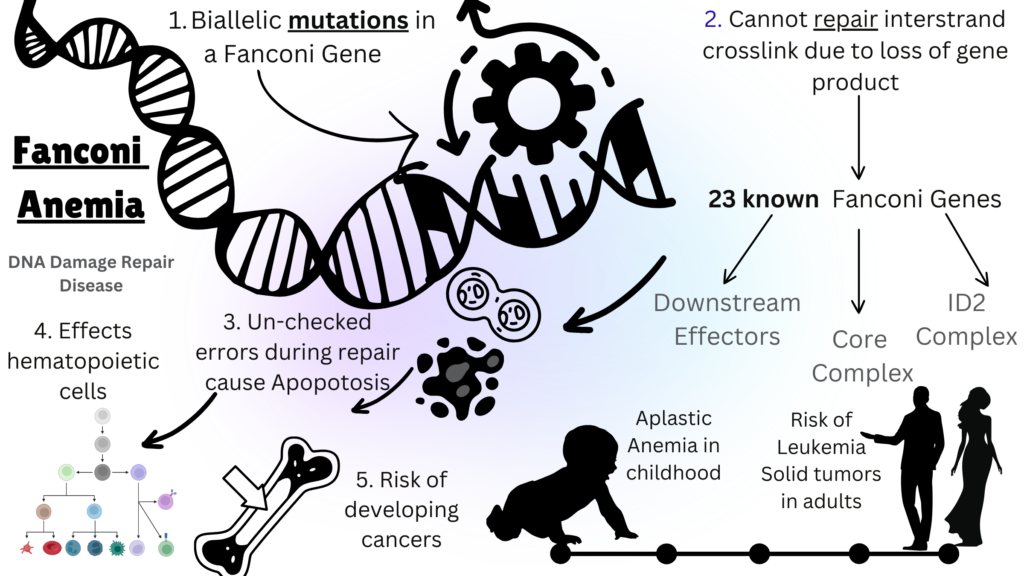
Image Source: Malini Gupta
Diagnosis and Current Treatment
Fanconi anemia (FA) is typically diagnosed before age 12, but symptoms can appear in adulthood. Prenatal diagnosis can be performed with a chromosomal breakage assay on fetal blood or by genetic testing if a mutation is known. Bone marrow failure (BMF) is a serious complication due to impaired DNA repair in bone marrow stem cells, leading to anemia, infections, and leukemia. Supportive care includes RBC transfusions, hematopoietic stem cell transplantation (HSCT) is the only curative treatment, replacing defective stem cells with healthy ones. Androgen therapy can improve blood counts but carries liver toxicity risks and does not reduce leukemia risk. Regular screening for hematological malignancies and solid tumors is recommended, especially in adolescence and after transplantation, due to increased cancer sensitivity from chronic graft-versus-host disease (GVHD) Regular screening for hematological malignancies is advised during childhood for non-transplanted patients. The detection of a clonal event, except for 1q anomalies, usually lead to consideration of transplantation.
For more on the latest curatives for this disease, stay tuned for F for Fanconi Anemia part II. Meanwhile, click here for more available resources and support groups for Fanconi Anemia.
Dr. Malini Gupta, Ph.D.
Sources

