Crigler-Najjar Syndrome (CNS) is a rare, autosomal inherited disorder characterized by a loss or deficiency of the liver enzyme Uridine Diphosphate glucuronosyltransferase (UGT), resulting in the inability to convert and clear bilirubin from the liver. Bilirubin is an orange-yellow pigment produced by the normal breakdown of worn-out RBCs, by a process called hemolysis. To be converted into a harmless soluble form, by a process called “Bilirubin conjugation”, UGT is required for glucuronidation of unconjugated bilirubin, for it to be excreted through the bile and into the intestines. Depending upon the mutations in the UGT1A1 gene, it can lead to either a complete absence/ inactivity of UGT (Type I CNS) or severely reduced (Type II CNS). Type I CNS is characterized by complete inactivity of UGT, which can be fatal and may lead to childhood mortality whereas Type II has reduced activity of UGT and can be managed by medical interventions.
The build-up of free or unconjugated bilirubin results in jaundice, which can reach up to the brain and causing “Kernicterus”, a severe form of brain damage seen mostly in infants. In some cases, individuals with CNS type II, may not develop kernicterus until later in childhood or in early adulthood. In CNS type II, some people are not diagnosed until adulthood. Kernicterus is rare in Crigler-Najjar syndrome type II but can occur especially when an affected individual is sick, not eating, or under anesthesia.
Orphanet Description and Clinical Features
| Classification Level | Disorder |
| Worldwide Prevalence | 1-9/1000,000 |
| Inheritance | Autosomal recessive |
| Age of Onset | Infancy, Neonatal |
- Severe jaundice shortly after birth may raise suspicion of Crigler-Najjar syndrome, presented with isolated jaundice (neonatal jaundice): yellowing of the skin, mucous membranes, and whites of the eyes, being more severe in CNS type I than CNS type II.
- Kernicterus or bilirubin encephalopathy with hypertonia, deafness, oculomotor palsy, lethargy, vomiting, fever, and/or unsatisfactory feedings.
- Other symptoms that may follow include the absence of certain reflexes; mild to severe muscle spasms, including spasms in which the head and heels are bent or arched backward and the body bows forward (opisthotonus); and/or uncontrolled involuntary muscle movements (spasticity).
- In addition, affected infants may suck or nurse weakly, develop a high-pitched cry, and/or exhibit diminished muscle tone (hypotonia), resulting in abnormal “floppiness.”
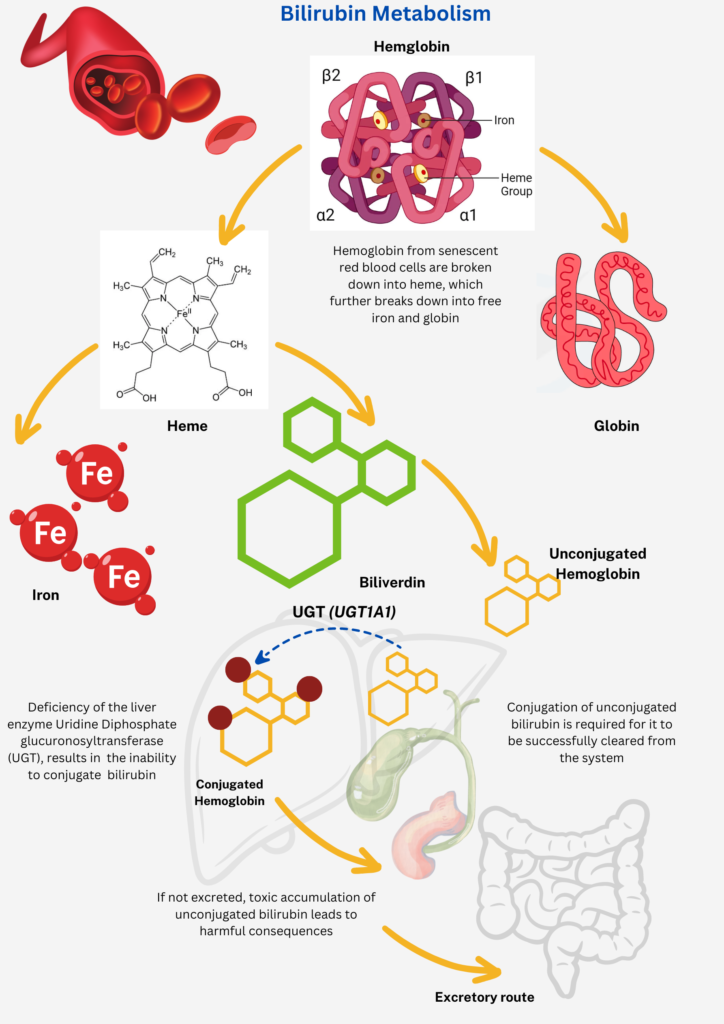
Image Source: Malini Gupta
Diagnosis and Current Treatment
- Physical examination reveals isolated jaundice, while lab tests show markedly elevated unconjugated bilirubin levels alongside normal liver function. Abdominal imaging (such as CT scans or ultrasonograms) usually appears normal.
- Presently, the definitive diagnosis relies on genomic DNA analysis, eliminating the necessity for liver biopsy.
- Genetic testing targeting mutations in the UGT1A1 gene can also validate the diagnosis.
- Differential diagnosis depends on the type of CNS including disorders of excessive bilirubin production (hemolysis) and mild hepatic deficiency of hepatic UGT1A1 (Gilbert syndrome).
Treatment for Crigler-Najjar syndrome type I involves hospital-administered phototherapy followed by home treatment for 10-12 hours daily. Bilirubin chelators may also be prescribed. Type II patients may receive daily phenobarbital, though suitability varies. Gene therapy for severe type I cases is experimental, undergoing clinical trials. June 21st is Crigler-Najjar Day, highlighting light’s importance, especially for affected children, as the longest day of the year.
For more on the latest curatives for this disease, stay tuned for C for Crigler-Najjar Syndrome part II. Meanwhile, click here for more available resources and support groups for Crigler Najjar Syndrome from around the world.
Dr. Malini Gupta, Ph.D.
Sources

