Acromegaly is a rare, acquired and slowly progressive condition caused by excessive production of growth hormone (GH) by the pituitary gland, due to the presence of a “pituitary adenoma”, a benign tumor. The pituitary gland is a pea-sized endocrine gland located at the base of the skull, in charge of producing several essential hormones and releasing them upon bodily requirements. Gigantism and Acromegaly usually result from the timing of over-secretion of GH, if it happens in childhood it results in Gigantism whereas if happens later in life, it leads to Acromegaly The disease doesn’t have a known genetic cause yet. Although usually related to pituitary adenoma, in very rare cases Acromegaly may also be caused by ectopic secretion of growth hormone-releasing hormone (GHRH) which can be blamed for pituitary hyperplasia. AIP or aryl hydrocarbon receptor-interacting protein has been deemed to be one of the determining factors for childhood or adult onset of this disease. Acromegaly can also be part of multiple endocrine neoplasia syndromes MEN1, Carney complex, or Familial isolated pituitary adenoma (FIPA), or even very rarely occur as secondary to Xq26.3 chromosomal microduplications responsible for X-linked acrogigantism (XLAG), McCune-Albright syndrome. November 1st is celebrated as World Acromegaly Day.
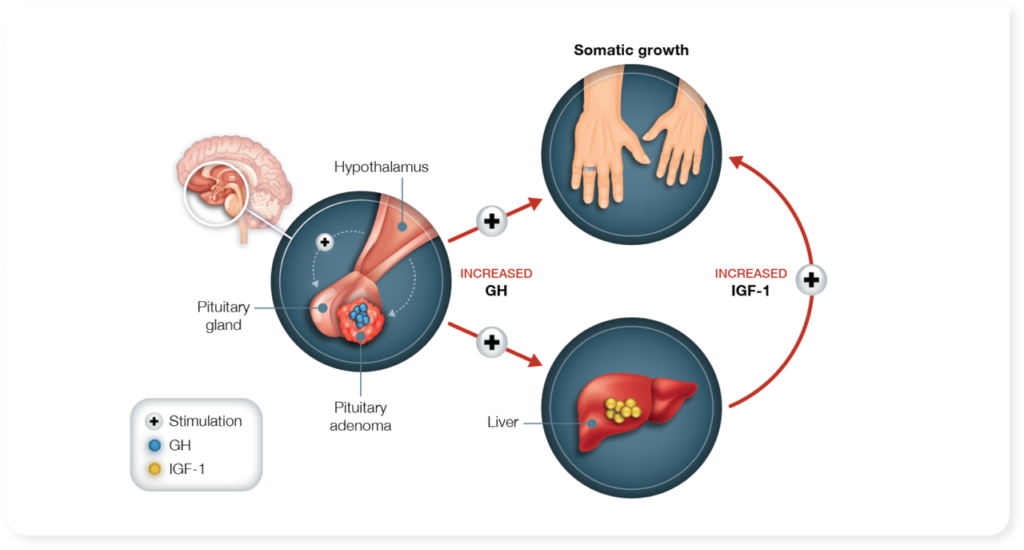
Image obtained from: https://signiforlar.com/hcp/acromegaly/acromegaly-today/unmet-need/
Orphanet Description and Clinical Features
- broadened extremities widened, thickened, and stubby fingers, thickened soft tissue,
- widened and thickened nose, prominent cheekbones, forehead bulges, thick lips, and marked facial lines.
- thickened forehead, and overlying skin, sometimes leading to frontal bossing.
- tendency towards mandibular overgrowth with prognathism, maxillary widening, tooth separation, and jaw malocclusion.
- rheumatologic, cardiovascular, respiratory, and metabolic consequences which determine its prognosis.
| Classification Level | Disorder |
| Worldwide Prevalence | 1/7500-1/35,800 |
| Age of Onset | Infancy, Childhood, Adolescent, Adult, Elderly |
| Annual Incidence | 1/91000 – 1/526,000 |
Diagnosis and Current Treatment
- Screening test for increased serum insulin-like growth factor I (IGF-I)
- Increased serum GH not suppressed by oral glucose tolerance test
- Assessment of tumor volume by imaging
- Echocardiography and sleep apnea testing
The first line of treatment is Transsphenoidal surgery – aiming at managing tumor compression by removing the lesion and bringing back the IGF and GH levels to normal. Medication such as dopamine agonists or somatostatin analogs may be recommended in addition to surgery. In cases where these medications don’t work, a GH antagonist called pegvisomant might be used. If medical treatments fail, radiotherapy could be considered as a last resort.
Adequate hormonal disease control is achieved in most cases allowing life expectancy akin to that of the general population however joint pains, deformities, and altered quality of life may still prevail. Due to its very slow progression, this disease is often diagnosed four to ten years after its onset making it mostly incident in adults (average 40-50 years). Uncontrolled GH levels and co-occurrence of diabetes and heart diseases reduced the patient’s life expectancy by as much as 10 years. Acromegaly treatment market trends have been seen to be focusing on early diagnosis and intervention, and biomarker discovery research to prevent tumor progression and mitigate long-term side effects of unregulated GH excess.

Image obtained from: “Day in My Shoes,”, released by Novartis in 2015 – https://globalgenes.org/blog/novartis-releases-acromegaly-infographic-entitled-a-day-in-my-shoes/
In the coming week, Malini will walk us through ongoing clinical trials and research organizations working on Acromegaly. Meanwhile, if you want to hear the voices of Acromegaly from around the world– you can mingle with the Acromegaly Community or reach out to Acromegaly Support.
Sources:

