

Sickle Cell Disease (SCD) is a group of rare genetic blood disorders characterized by the production of abnormal hemoglobin, known as hemoglobin S (HbS), which causes red blood cells to adopt a rigid, crescent or sickle shape. SCD manifests in different forms, and encompasses several related but distinct disorders, all of which are caused by genetic mutations in the hemoglobin gene. September is sickle cell awareness month. The main forms of SCD include:
Sickle Cell Anemia (HbSS): This is the most common and severe form of sickle cell disease, where an individual inherits two copies of the sickle cell gene (HbS), one from each parent. The presence of HbS in both hemoglobin chains leads to the formation of sickle-shaped red blood cells under low oxygen conditions.
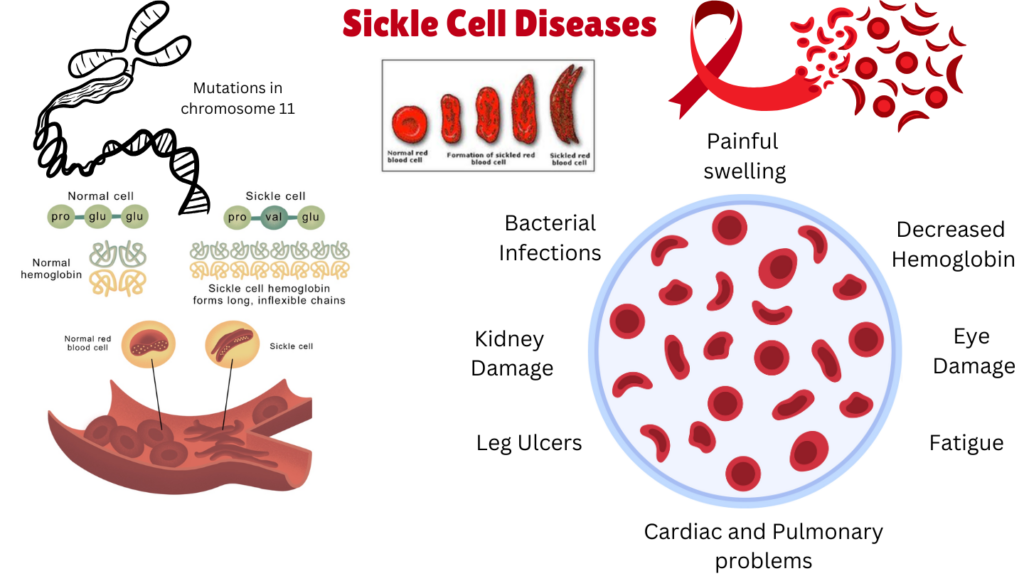
Symptoms are typically more severe and include frequent vaso-occlusive crises, organ damage, and a shortened life expectancy.
Sickle Cell Hemoglobin C Disease (HbSC):This form occurs when one copy of the sickle cell gene (HbS) is inherited from one parent, and the other parent passes on the hemoglobin C gene (HbC). Hemoglobin C causes red blood cells to become somewhat rigid but to a lesser extent than sickle hemoglobin.
Sickle Cell Beta-Thalassemia Disease (HbS/β-thalassemia): In this form, an individual inherits one sickle cell gene (HbS) and one beta-thalassemia gene. The severity of the disease depends on the type of beta-thalassemia mutation, as beta-thalassemia involves a deficiency in beta-globin production.
This form can vary in severity, ranging from mild to severe, and symptoms depend on the specific genetic mutation.
Clinical Description, classification and symptoms
| Classification Level | Disorder |
| Prevalence | 112/ 100,000 births |
| Age of Onset | All ages |
| Inheritance | Autosomal recessive |
The hallmark feature of sickle cell disease (SCD) is the obstruction of blood flow by sickled red blood cells, leading to various complications. Common symptoms include:
Pain Episodes (Vaso-Occlusive Crises): Severe, prolonged pain due to blocked blood flow.
Anemia: Chronic fatigue and weakness from rapid red blood cell destruction.
Jaundice: Yellowing of the skin and eyes from hemoglobin breakdown.
Infections: Increased risk, especially in children, due to spleen dysfunction.
Stroke and Organ Damage: Potential neurological and long-term organ damage.
Acute Chest Syndrome: A life-threatening condition affecting the lungs.
Diagnosis and Treatment Management
Diagnosis of sickle cell disease (SCD) is primarily done through newborn screening and hemoglobin electrophoresis, which identifies the presence of sickle hemoglobin (HbS). Genetic testing may also be used to confirm the diagnosis. Prognosis varies depending on the severity of the disease and the specific form of SCD, but with early diagnosis and proper management, many individuals can live into adulthood. While there is no universal cure for sickle cell disease, several management strategies can help alleviate symptoms and improve quality of life:
- Pain Management: Pain relief, often with opioid analgesics during a vaso-occlusive crisis, is a mainstay of therapy. Nonsteroidal anti-inflammatory drugs (NSAIDs) can be used for mild pain.
- Hydroxyurea: Hydroxyurea is a medication that can increase the production of fetal hemoglobin (HbF), a form of hemoglobin that does not sickle. This reduces the frequency of pain crises and other complications.
- Blood Transfusions: Regular blood transfusions can help maintain adequate oxygen delivery and reduce the frequency of sickle cell crises. Transfusions may also be used to treat acute complications such as stroke or acute chest syndrome.
- Bone Marrow or Stem Cell Transplantation: A hematopoietic stem cell transplant (HSCT), commonly known as a bone marrow transplant, is currently the only potential cure for SCD. However, it carries significant risks, including graft-versus-host disease (GVHD) and rejection.
- Gene Therapy: Gene therapy approaches, including CRISPR-Cas9 and lentiviral gene therapy, are promising newer treatments that aim to correct the genetic mutation causing sickle cell disease, either by repairing the HBB gene or by inducing the production of fetal hemoglobin. Clinical trials are ongoing.
- Vaccination and Prophylaxis: To prevent infections, particularly in children, individuals with SCD are routinely vaccinated against pneumococcus, meningococcus, and Haemophilus influenzae, and may also receive prophylactic antibiotics.
For more on therapeutics against this disease, stay tuned for S for Sickle Cell Disease Part II. Meanwhile, click here for more available resources and support groups for SCD. Read more on this rare disease here.
Sources
- https://rarediseases.org/rare-diseases/sickle-cell-disease/
- https://www.orpha.net/en/disease/detail/232?search=Sickle-cell-disease-and-related-diseases&mode=name
- Figure elements – http://www.sicklecellassociationofbc.com/about-sickle-cell.html
- Figure elements – https://sickle-cell.com/causes


