

Beta Thalassemia (BT) is an inherited autosomal rare recessive blood disorder caused by reduced levels of functional hemoglobin (Hb), the red iron heavy oxygen-carrying pigment of the blood. Hemoglobin is a tetramer of globin chains each bound to a heme group which is bound to iron which accommodates oxygen. The four types of globin chains – alpha (α), beta (β), gamma (γ), and delta (δ) combine in specific ratios to form HbF (α2, γ2), HbA (α2, β2) or HbA2(α2, δ2) types of hemoglobin. Point mutations in the β globin chain encoding HBB gene cause Beta Thalassemia. Inheritance of either one copy (minor) or both copies (intermedia or major) of this point mutation, results in either partial or complete deficiency of β chain synthesis respectively. While individuals with the minor form of this disease are often asymptomatic, individuals with BT major, inevitably require lifelong blood transfusions and clinical management. Alpha thalassemia is common in Southeast Asia, with high number of carriers in sub-Saharan Africa and the Western Pacific region whereas Beta thalassemia is most common among populations of Mediterranean, African and South Asian descent. The prognosis of BT although dependent on disease severity, usually is promising, if detected early and managed with clinical supervision.
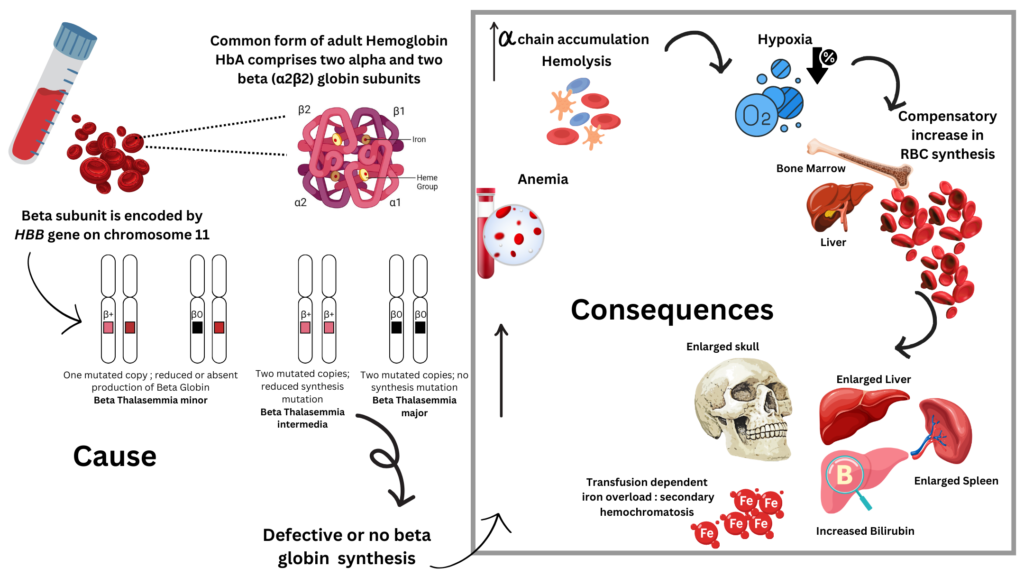
Image source: Malini Gupta
Orphanet Description and Clinical Features
| Classification Level | Disorder |
| Worldwide Prevalence | 1-9/100,000 |
| Inheritance | Autosomal dominant Autosomal recessive |
| Age of Onset | Infancy, Childhood, |
| Annual Incidence | estimated at 1/100,000 |
- Onset usually happens early; from 6-24 months of age.
- Thalassemia minor (BT-minor, BT trait) is a heterozygous form; usually asymptomatic.
- Thalassemia intermedia (BTI) patients may or may not require frequent blood transfusions as the anemia is less severe and diagnosed later in life compared to BT-major. But hypersplenism, cholelithiasis, extramedullary hematopoiesis, thrombotic complications and progressive iron overload are the main clinical features that may complicate the course of BTI.
- Thalassemia major (Cooley anemia; BT-major) is homozygous and often involves splenomegaly and microcytic and hypochromic anemia because of dyserythropoiesis and hemolysis.
- Severe anemia demands systematic transfusions to maintain normal hemoglobin levels (90-100 g/L) to allow normal activity.
- Complications from long-term repeated blood transfusions result in iron overload hampering vital prognosis (mainly due to cardiac involvement) and causing significant morbidity (due to endocrinal and hepatic iron deposition).
- BT trait can be associated with trichothiodystrophy or X-linked thrombocytopenia in rare instances.
Diagnosis and Current Treatment
- Prenatal diagnosis available via genetic counseling based on more than 200 identified mutations β0 or β+
- Analysis of Hb by electrophoresis or HPLC
- In BT-minor, the levels of HbA2 are increased and the levels Hb are usually normal to low with microcytosis and hypochromia.
- BT-major: HbA is absent or greatly reduced and HbF predominates.
Treatment options traditionally involve a combination of regular blood transfusions along with iron chelation therapy to reduce transfusion-mediated iron overload complications. A promising treatment for BT major involves hematopoietic stem cell transplantation in children. The latest in the line of treatment happens to be gene therapy.
For more on the latest curatives for this disease, stay tuned for B for Beta Thalassemia part II. Meanwhile, if you want to hear the voices of Beta Thalasemmia from around the world – you can check Life with Beta Thalassemia, Thalassemia International Foundation, Challenge TDT or Cooley’s Anemia.
Sources
- https://www.orpha.net/en/disease/detail/848
- https://my.clevelandclinic.org/health/diseases/23574-beta-thalassemia
- https://rarediseases.org/rare-diseases/thalassemia-major/


